A Lazy Approach To Problem Solving
Solve the right problem, not the given problem

Leon Redbone is a singer and guitarist specializing in interpretations of early 20th-century music—including jazz, blues, and Tin Pan Alley classics. With his own unique style, he has an almost cult-like following—to which I belong. One of his classic renditions is of the song “Lazy Bones” written back in 1933 by Johnny Mercer and Hoagy Carmichael.
Today, inspired by the song, I would like to suggest that we actively be more lazy—that we be lazy when it comes to problem solving.
The central thesis is: Do not blindly solve a problem by working on the problem. Instead ask, is the problem the real fundamental question? Or is there a more basic—and hopefully easier to solve—problem that needs to be solved?
The idea taken to its extreme would be to not solve the problem at all, but continue on as if the problem had been solved. Only solve the problem when you are forced to. This is related to the notion in programming language theory called lazy evaluation. Its history is a bit convoluted. The notion was introduced into the mother of all languages, the lambda calculus, by Christopher Wadsworth in 1971, then for other programming languages in two independent papers in 1976 by Peter Henderson and Jim Morris, and by Daniel Friedman and David Wise.
In order to explain the notion I will give three examples. The first is almost trivial, the second is almost impossible, and the third is just right, well at least the third seems to be interesting and new.
Read on to see what we are talking about. To me it sounds a bit like the story of Goldilocks and the Three Bears. I recall that one bed was too small, one too large, and one was just right. Here one example is too easy, one is too hard, and one is approachable.
Trivial Example
I have discussed the issue of problem solving and talked about the “A to B to C fallacy.” There I also talked about solving the right problem. Here is the promised trivial example, which hopefully will help demonstrate the idea of being lazy. Imagine that I present to you a large-degree polynomial 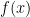

which I promise has only real roots. That restriction is not necessary, but will make the example easier to explain. If I ask you for the roots 
The lazy approach is to immediately say “sure, I have the roots.” Just pretend that you have them and call them
ch works extremely well.
Note, this is based on the formula

together with 

Impossible Example?
The above example, while trivial, is one that I have thought about as a motivation for trying to be lazy with other more important problems. One of the central problems in biology is the problem of discovering the structure of a protein–the protein folding problem. The standard dogma is that proteins fold into a 3-dimensional structure, and that this structure holds the key for understanding how the protein works.

However, there is some recent disagreement that we will discuss another day. Unfortunately, discovering the 3-dimensional structure is still a challenging and difficult problem.
I have often asked biologists, why do they want this structure? One paper raises this very question and answers it this way:
- Structure is more easily conserved than sequence.
- Similar folds often share similar function.
- Remote similarities may only be detectable at structure level.
Another reason for their interest in finding folded structures is that they hold the key for where small molecules can “dock” and in this way turn off the protein. If the protein causes a disease, then stopping its function can be a cure. Essentially, these small molecules are drugs. The idea is if we can determine the structure, then we can in principle find which small molecules can dock, and thus we can find new drugs.
The lazy problem-solving approach here says: Given a protein, do not determine its structure, but rather determine which small molecules can dock. I have discussed this over the years with my colleagues in biology. The usual answer is, with an amazed look: “can you do that?” Unfortunately, today I cannot. But perhaps, a big perhaps, perhaps we can determine potential molecules that can dock without the computational effort of finding the structure.
You can see, I hope, the analogy with the root-finding problem.
Do not find the roots (the structure); instead find the docking molecules (the norm of the roots).
I doubt it will be as simple for proteins as for polynomials, but I do not believe that this is an avenue that people are currently addressing. If the lazy way can be followed up, then here is a new approach to the whole drug discovery problem.
Possible Example
Let’s turn to a problem that lies between the previous two. It does not seem trivial, but is possible. The idea is that when we solve a system

for 
The answer is yes, provided we can use pre-processing. In particular we can prove the following theorem:
Theorem 1 Suppose that
is an
by
be fixed, and let
(a set of indices) be fixed. Then there is a randomized polynomial time algorithm running in time
that given the vector
determines
up to a multiplicative error of
, where
is the solution to
. The algorithm does require pre-processing time that is
given
, and

Note that this theorem is exactly analogous to the polynomial example. We can compute partial information about the solution 
- The matrix stays the same, but the right hand side,
, changes.
- Only partial information about
- There is no (obvious) way to invert
I would also like to add that a similar theorem can be proved when other functions of
Open Problems
The song “Lazy Bones” is also a brilliant example of problem solving. The legend, which appears to be true, is that it happened as follows. According to Carmichael, in an interview, Mercer came into Carmichael’s apartment in New York one day and:
[He] saw Hoagy “snoozin” on his couch. Mercer said, “Hoag, I’m gonna write a song called ‘Lazy Bones’.” Carmichael said, “Well, let’s get at it.” They went over to Hoagy’s piano, Johnny said the first line and Hoagy started playing a melody. The song was done in twenty minutes.
Perhaps we can solve more problems with this let’s-get-it-done-now attitude. The next time you see a colleague asleep on a couch, say “let’s solve 
[Added third “critical property” and changed formatting at end.]
Trackbacks
- Mathblogging.org Weekly Picks « Mathblogging.org — the Blog
- Michigan Coding, Complexity, and Sparsity Workshop « Gödel’s Lost Letter and P=NP
- One Man’s Floor Is Another Man’s Ceiling « Gödel’s Lost Letter and P=NP
- Cancellation is a Pain | Gödel's Lost Letter and P=NP
- Quantum Switch-Em | Gödel's Lost Letter and P=NP


Wouldn’t the lazy approach be to assume P != NP and tackle something else?
Well.. everybody does not have to work on P=?NP problem.
Reblogged this on Room 196, Hilbert's Hotel.
Dear Professor Lipton:
For the Ax = b problem, will your technique extend to find the l_infinity norm (rather than l_2 norm) of x in time O(n log n/e) or even O(n^2 log n/e)? It seems such a result would be interesting since it may provide a significantly faster way to implement page-ranking algorithm.
Ever since the idea I gained an advanced understanding of the idea of the certificate and some intricate details of asymptotic notation, I have thought of similar questions.
Ideas that have crossed my mind:
– Get the best (non-asymptotical) SAT solver there is out there, perhaps tailored to your own problem. Then solve a single instance that is worth solving: e.g. solve TSP for a state or a medium-to-small country. Even easier: solve the CSP for loading a vehicle. The idea is that even if the exponential time hypothesis is true, for relatively small input sizes and given our (ever?) increasing computational power, we can solve an instance that really matters. Even if it takes months, once we got the result,we are covered for quite some years (until the road network changes, that can be half a century, or a new vehicle is introduced).
-In a similar notion: Inspired by Aaronson’s call for experiments on computing the permanent, let’s do the same for other problems. Let’s imitate the other great “theoretical” science, theoretical physics. If we had a supercomputer with funding equivalent of those of LHC, we could probably gain some insight by solving, say, all SAT instances for n=50 or n=100.
– Suppose that the exponential time hypothesis exists. However, by running the experiments on the TCS-LHC I mentioned before , we find out that for every n, there is a “kernel” instances that acts as a “basis” for space of all instances. Furthermore, there is a $n^{logn}$ algorithm for reducing every other instance to one of those instances. By precomputing whether those instances are satisfiable or not up to a reasonable input size, we could live in a world where P=NP is almost true. You could also consider a variation where the kernel is always satisfiable, thus having a good algorithm for SAT (of course in this case the ETH is false). This also led me to the idea that a polynomial-sized number of steps into transforming co-SAT to the constant 0 formula (that all unsatisfiable formulas are isomorphic to) could serve as a certificate. Again, the main idea is not to try to solve the formula but rather circumvent the problem, in a way.
As a disclaimer, those are rather vague thoughts than research proposals. If done carefully, I believe #1 can improve our lives in a significant way and could be our only hope for exact solutions if P is not equal to NP. In a perfect world where budget is not a problem, #2 should produce at least some results. As for #3 , I believe it requires a deep understanding of the problem.
Not a case of laziness but this reminds of homomorphic encryption where the tally of a vote can be decrypted without decrypting individual votes. In this case it’s not a matter of being lazy and saving on computation, but of preserving privacy by hiding intermediate steps.
“Do not find the roots (the structure); instead find the docking molecules (the norm of the roots). ”
Oog, the asymmetry hurts me. Maybe reverse one of the clauses?
Hi Dick, I am embarrassed to say I was unable to figure out the algorithm for finding the norm of the roots of the polynomial even with your clue for the quadratic sum. Could you (or anyone who wishes to help) hint at any intermediate steps? I am assuming that the result is elementary to work out (perhaps, this is incorrect and some standard textbook result is employed). Thanks as always for presenting these fun little nuggets.
The norm is the square root of the sum of the squares. So once you have the sum of the squares you are there.
The important point is that the sum of the squares is a symmetric function of the roots. A theorem/algorithm of Gauss allows you to express any symmetric function as a polynomial in elementary symmetric functions (the cofficients of the polynomial, up to sign). Dick’s example is a specific one: $\sum_i x_i$ is $-a_{n-1}$ and $\sum_{i < j} x_i x_j$ is $a_{n-2}$.
Thanks so much, Dick, Victor, based on Victor’s hint I found this reference http://www.danielmathews.info/articles/games_with_galois.pdf which expands the algorithm that Victor mentioned. An elementary yet fun fact.
There is a passage in Jorge Luis Borges’ story Averroes’s Search (a story that is strikingly popular among scientists) that suggests natural broadening of the idea of lazy research strategies:
• … because we’re lazy?
• … because we’re timid?
• … because we’re aspect blind?
• … because we’re prudent?
• … because the creation is unitary?
• … because the creator is merciful?
As usual in Borges’ stories, one has to decide for oneself the relative weight and appropriate context of each of these readings.
With regard to dynamical-versus-structural modeling in synthetic biology, at the seminars that I attend, *that* conflict is pretty much over — structure is invariably the starting-point for dynamical modeling. The paradoxical result is that nowadays the demand for observational coarse-grained initial structure is burgeoning (that is, initial structures having spatial resolution of order 0.5 nanometer and larger), while finer-grained structure is conceived as dynamical-yet-simulable with modern molecular dynamics codes.
This provides a modern context for Feynman’s lazy-researcher advice in his celebrated essay There’s Plenty of Room at the Bottom: (1958)
And here Feynman is not talking about making biology a little bit easier … he is talking about making biology many orders of magnitude easier&nbbsp;… a goal that is unsurprising given Feynman’s research focus at the time. This is truly a lazy researcher’s vision! 🙂
As with Feynman’s pioneering vision of computing, and Feynman’s pioneering vision of nanotechnology, nowadays we are making remarkable progress with Feynman’s pioneering vision of microscopy — and it is striking that the mathematical tools, physics insights, engineering technologies, and enterprise cultures of these three Feynman visions are dovetailing very naturally … very much as Feyman so clearly foresaw. Rapid advances in Feynman-style computational simulation methods, as applied to Feynman-scale biomolecular structures, as imaged with Feynman-resolution microscopes, now are bringing to fruition a 21st century that fulfills the main narrative theme of all of Feynman’s stories and lectures — a narrative theme that Feynman invariably developed with care and attention to detail as scrupulous as Borges’ — This is going to be TERRIFIC! 🙂
Another natural link between Dick’s theme of “A Lazy Approach To Problem Solving” and Borges’ story Averroes’s Search is suggested by the Afterword that Borges provides:
Similarly, with regard to the main challenges of STEM in general (and FTQC in particular):
Borges’ “lazy” response to his intractable problem was to write an immortal short story. Similarly, by “lazily” doing whatever we can, with the tools that we have at hand, perhaps we of the 21st century STEM enterprise can make progress too.
Let me mention two related areas from TCS: Streaming algorithms and in particular the work of Noga Alon, Yossi Matias, and Mario Szegedy (which the first and last examples resemble), and constant-time algorithms like those in property testing.
Highly relevant to Dick and Ken’s topic of “Lazy Approaches to Problem Solving” is economist Tyler Cowen’s much-read, oft-praised, and sometimes-disdained recent book
In regard to Prof. Cowen’s “Great Stagnation” thesis, some folks are motivated to study mathematics as humanity’s single most capable tool for avoiding a planetary-scale “Great Stagnation” … other folks study mathematics as a Gödelian/Platonic realm in which earthly questions of stagnation are utterly irrelevant … and the careers of a great many eminent researchers (Claude Shannon? John von Neumann? Bill Thurston? Jim Simons?) illustrate hybrid research strategies of one variety or another.
Regardless of one’s personal inclination, Cowen argues (in effect) that every STEM researcher should ask-and-answer: “Is *my* discipline at-risk of a Great Stagnation?”
What’s distressing about the present era — and constitutes a strong sign (IMHO) that STEM stagnation is ubiquitous — is that the younger the researcher, the greater the prevalence of the answer “yes”. 🙁 🙁 🙁
Can Dick and Ken’s “lazy approach” to problem-solving help us to find more Tyler Cowen’s “low-hanging fruit” … and thus create the abundance of career opportunities for young mathematicians, scientists, and engineers, upon which the long-term viability of the STEM enterprise depends? It seems (to me) that the answer is “yes, definitely”
Just a quick thought. I think the proposal is to identify geometric properties of the proteins and the docking molecules such that the problem can be reduced to one of identifying group structures. Presumably if one can enumerate a list of proteins and molecules on can make a matrix and identify which cases are known to be compatible.
The question then is how to parameterize the geometry of the proteins and molecules. Clearly in some cases a molecule will see a hole it can fill and in others it will not. Presumably the problem can be reduced down to some type of eigenvalue problem. I suppose there is a lot a ground work already in this area, but I agree with the general thesis that there should be a way to parameterize without identifying the actual folding structure of the protein.
Hal, it turns out that structural biologists compete among themselves to predict protein structures using very much the same competiton rules that (1) chess-players use (both human and computer) and (3) crossword-puzzle solvers use (both human and computrer), and (3) SAT-solvers use (computer only!).
Among the best-known of these contests is Critical Analysis of Structure Prediction (CASP), which is held every two years … entries for CASP10 are now being solicited.
The mathematical and algorithmic basis for structure prediction (which nowadays has become astoundingly successful!) is covered in texts like Frenkel and Smit Understanding Molecular Simulation: from Algorithms to Applications.
Rather like computer chess, as the simulation of smaller-scale molecular systems in synthetic biology has become tractable — in consequence of Moore’s Law improvements in computing hardware, simulation algorithms, and structural microscopy — the focus is shifting to dynamical and epimolecular structural relations.
This is a burgeoning field that offers terrific career and enterprise opportunities for young researchers of every persuasion.
Also, in reference to the ongoing Kalai/Harrow debate, it is hugely stimulating to study the CASP summary reviews side-by-side with a wonderful preprint this week, out of Alán Aspuru-Guzik’s group, titled Simulating chemistry efficiently on fault-tolerant quantum computers (arXiv:1204.0567). This preprint
works through the nuts-and-bolts of designing an FTQC to estimate (for example) compute ground-state energies of lithium hydride (LiH, a two-nucleus system).
One take-home message is that at present rates of progress it will be very many decades — plausibly never? — before FTQC hardware can tackle CASP-scale systems of many tens of thousands of interacting, thermalized electrons and nuclei.
And yet research in FTQC *already* exerts a hugely beneficial effect on CASP-class enterprises, partly by steadily enlarging the scope of dynamical systems known to be simulable by classical methods, and partly by steadily improving the sensing and channel capacity of the instruments (mainly x-ray interferometers and magnetic resonance sensors) that feed the CASP community’s insatiable demand for starting molecular structures.
So we don’t have to wait decades to realize immense benefits from fundamental FTQC research … those benefits are being realized right now.
John, thanks for the links. I was wondering if you knew anything more about topological nomenclature associated with protein folds. I found the following reference that has a nomenclature that can capture all the information on the protein folds. Since the paper is over 10 yrs old I didn’t know if it contains standard nomenclature.
http://peds.oxfordjournals.org/content/11/9/723.full.pdf
I noticed that protein data bank data provides data based on 3d coordinates
http://en.wikipedia.org/wiki/Protein_Data_Bank_%28file_format%29
I don’t have time to dig into how the simulation models are built for the CASP website, but I am curious if they translate structure into some standard topological nomenclature.
Hal, from a formal point of view, finite-temperature systems necessarily exhibit dynamical structure … and biological molecules definitely are finite-temperature systems.
So the short answer to your question is that the primary outputs of molecular simulations nowadays are *not* short topological descriptions of structure, but rather huge files (terabytes) of simulated trajectories. These trajectories encode the answer to practical questions like “Which residue should we modify to increase the binding affinity of this protein with respect to some target molecule?”
Thus, as the domain of systems and synthetic biology enlarges from genomics to epigenomics, what once was aptly called “structural biology” now perhaps might better be described as “dynamical biology”.
Thanks John, it seems that the interest then isn’t just in the minimum energy folded state but also the path, My question was more motivated by the idea that the final state was of greater interest, and the thought was whether or not there is a linear nomenclature that could completely capture all the structure information of the protein.
If one accepts Anfinsen’s Dogma, then there is some unique native structure determined solely by the amino acid sequence. If the structure nomenclature and the sequence nomenclature are sufficient to completely capture the information, then all one has are two different basis describing the same information.
If s represents a sequence and t represents a structure we could say the following (taking liberties with indices):
There exists an operation A that for a sequence transition i to j we can write:
A_ij s_i = s_j
and there exists an operation B such that for a basis change from s_j to t_j we can write:
B_jj s_j = t_j
and there is an operation C such that
A_ij B_jj = C_ij
and
C_ij s_i = t_j
so that in a nice idealized world one could take information about a known sequence and use it to determine the structure of some other sequence. This seems to be called ab initio prediction.
What would be interesting, would be to take some sequence with some feature (we’ll call it a wavelet) and propogate it up and down the length of the sequence to see what happens to the strucure.
I just have a hunch that there would be some simple set of rules for certain proteins but that as the problem gets more complex, the transformation rules would as well (much like turbulence).
However, if the trajectories themselves are important then this sort of approach is obviously limited.
As for PvsNP, it doesn’t seem to be the right question to ask. IMHO, it doesn’t make sense to prove facts about algorithms that no computer will ever run. The real issue is more about the future: will it always be hard to break RSA? The existence or inexistence of efficient algorithms is often less interesting than our ability or inability to find them.